Separating the Wheat from the Chaff: AI in practice: can we bridge Language Models and Intelligence Engines to go further, faster?
Over my two decades in computational biology and AI-driven research, I’ve seen firsthand how both omics-based machine learning and LLM-driven literature mining have reshaped drug discovery. Early in my career, I remember running my first Random Forest Classifier on RNA-seq data and being amazed by the patterns that emerged. Yet I also experienced the frustration of reading dozens of papers manually, hunting for clues. In this article – the first of a series – I share insights from our own projects to clarify these complementary paradigms, explain why they’re often conflated, and guide you in choosing and combining ML and LLMs for your research goals.
Machine Learning on Primary Omics Data
At its core, ML on omics data is a data-driven, evidence-based approach. You begin with raw measurements—gene expression counts, mass spectrometry intensities, or single-cell sequencing reads—and use algorithms to discover patterns that distinguish experimental conditions, predict phenotypes, or identify potential biomarkers around a given question or questions. Key characteristics include:
- Structured input: Numeric matrices with samples as rows and molecular features as columns.
- Statistical rigour: Emphasis on normalisation, batch correction, feature selection, and cross-validation to avoid overfitting and false discovery
- Biological hypothesis testing: Often framed around “Is gene X differentially expressed?” or “Can we predict drug response from mutational profiles?”
- Model interpretability: Methods such as Random Forest, support vector machines, or explainable deep learning architectures provide feature importance measures that map back to genes or pathways.
While ML on omics can unearth novel molecular signatures and generate hypotheses directly from experimental data, it requires careful experimental design, sufficient sample sizes, and domain-specific preprocessing pipelines. If used correctly, however, ML has the potential to reveal fundamental mechanisms underlying disease.
LLM-driven Literature Analysis
In contrast, LLMs such as GPT-4, BioBERT, and PubMedGPT operate on unstructured text—titles, abstracts, full-text articles—and are trained to capture linguistic and semantic patterns across millions of documents. Their primary applications in biology include:
- Knowledge retrieval: Summarizing what is known about a gene, protein, or pathway.
- Hypothesis generation: Proposing novel connections by analogical reasoning across studies (e.g., linking a drug’s mechanism in oncology to potential applications in immunology).
- Automated annotation: Tagging entities (genes, diseases, compounds) and extracting relations (e.g., “Gene A inhibits protein B”).
- Literature review assistance: Drafting outlines, identifying key papers, and suggesting experimental designs based on historical precedent.
These models excel at synthesizing prior knowledge across thousands of publications, but they do not ingest raw experimental measurements—LLMs know what has been published, not what your sequencer measured.
Why the Confusion?
Several factors contribute to conflating these distinct approaches:
- Hype and marketing: “AI for biology” is often presented as a monolith, obscuring the diverse subfields it encompasses.
- Shared terminology: Both use “machine learning,” “deep learning,” and “neural networks,” leading non-experts to assume they solve the same problems.
- Integrated platforms: Some vendors bundle omics analytics with literature mining in a single interface, further blurring boundaries.
- Overlap in outputs: Both approaches can suggest biological mechanisms or candidate targets, but the provenance of those suggestions is fundamentally different.
Problems with Literature Bias in LLM-Driven Discovery
While LLMs offer unprecedented scale in scanning and synthesizing publications, they inherit—and can even amplify—biases present in the underlying literature. Key issues include:
- Publication bias: Positive results and novel mechanisms are far more likely to be published than null or “negative” findings. An LLM trained on these texts will disproportionately surface hypotheses that align with what made it to print, potentially overlooking dead ends or contradictory data.
- Citation bias: Highly cited articles exert outsized influence on the model’s internal representations. Seminal but outdated papers may dominate, while newer or niche studies remain under-represented.
- Language and geographic bias: English-language journals and research from North America and Western Europe dominate global publishing. Important findings from non-English or lower-resource settings may be marginalised, skewing the LLM’s view of the field.
- Temporal bias (“knowledge decay”): LLMs trained on literature up to a certain cutoff date won’t “know” the latest breakthroughs. This can mislead users into pursuing directions that have already been refuted or superseded.
- Reinforcement of dogma: As LLMs suggest links based on frequency of co-occurrence (e.g., “p53” and “apoptosis”), they can reinforce prevailing paradigms and discourage exploration of under-studied pathways.
Implications:
- False confidence: Researchers may take LLM-generated hypotheses at face value without scrutinising whether they are artefacts of bias.
- Resource misallocation: Time and funding can be wasted chasing “discoveries” that are merely echoes of publication trends.
- Ethical and equity concerns: Under-representation of certain diseases or populations in the literature can perpetuate health disparities when LLMs guide research investment.
Addressing these biases requires both awareness and methodological safeguards—mixing literature-mining with data-driven validation (see next section), applying debiasing strategies (e.g., weighting under-represented studies), and keeping the models’ training cutoffs transparent.
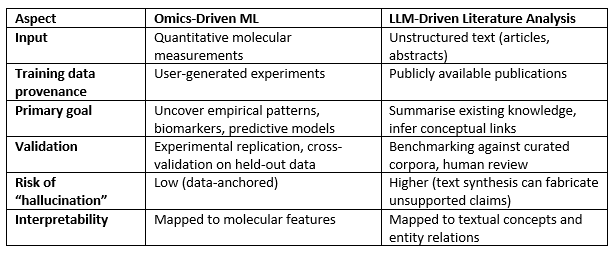
Key Distinctions at a Glance
Bridging the Gap: Synergy, Not Substitution
Rather than choosing one approach over the other, the real power lies in the appropriate combination of ML and LLM techniques:
- Evidence-based discovery: START with application of ML on your omics data to discover the underlying causal biology of disease, identifying possible targets in disease pathways.
- Hypothesis refinement: Use LLMs to scan the literature and identify any corresponding (or even contradictory) hypotheses concerning pathway involvement or drug repurposing, whilst remaining vigilant to bias.
- Data-driven validation: revisit application of ML on your omics datasets to augment those hypotheses, old and new, identifying statistically robust signals.
- Iterative learning: Feeds back experimental results—validated biomarkers or pathway modulations—into literature-based workflows to uncover mechanistic explanations or overlooked studies.
We can illustrate this interplay between ML and LLM: for example, imagine that output from our initial ML analysis of transcriptomic data in a tumour population indicates that there are multiple subtypes of a disease, as is often the case. The enriched gene product features specific to subtypes but varying over the whole population might then be used as inputs in an LLM to identify their known biological roles in pathways and biological processes from the literature and other knowledge bases.
These hypotheses can then be fed back to the ML model, resulting in enrichment of the molecular landscape around the pathways of interest based on ML evidence in the different subpopulations under investigation. This augmented list is then run through ML-based network inference analyses to model the pathway system, determine its drivers and investigating how individuals vary in their network behaviour. Running this across multiple subpopulations builds a digital twin of the pathway – which in turn can also be interrogated using ML to build companion diagnostic models.
Ultimately, disease drivers identified within the ML models can be fed back into the LLM to determine their known druggability, to identify pharmacophores and to identify small molecules that might be suitable drugs through binding into the gene product, ranked using Lipinski’s rule of five and other metrics pertinent to the disease in question.
In this way, the strengths of ML and AI are utilised to their full extent: The ML uses evidence-based approaches to extract contextualised knowledge based on robust evidence from primary data in an explainable fashion, and these results are fed into the knowledge-based expert in the form of an LLM in order to understand and integrate the findings in the current knowledge base.
Conclusion
Machine learning on primary omics data and large language model–driven literature mining are complementary, yet fundamentally different, tools in the modern biologist’s toolkit. Properly distinguishing between data-first discovery and text-first synthesis—and accounting for literature biases—ensures that research programs are designed with realistic expectations, robust validation strategies, and optimised resource allocation. By embracing both approaches in a coordinated workflow, scientists can accelerate the journey from raw data to mechanistic insight—and ultimately to transformative therapies and diagnostics.
About the Author
Professor Graham Ball, PhD, co-founder and CSO of Intelligent OMICS, brings over 20 years of hands on experience blending omics analytics with AI-driven literature discovery. Prof Ball leads interdisciplinary teams that transform multi omics data into clinically relevant insights.
This is the first article in a series of pieces written by Professor Ball. The series culminates in an ML and AI Training seminar for the OBN network in Q1 2026.

Intellomx contact information
Simon Haworth, CEO: simon.haworth@intellomx.com
General enquiries: info@intellomx.com
Website: www.intellomx.com
Intellomx (Intelligent OMICS Ltd), registered in England company number 06500709


